Read alignment and visualization using JBrowse2
This tutorial shows how to go from a set of fastq files, perform alignment and then visualize the alignments relative to genome annotations using JBrowse2.
We start with activating our conda environment (adding channels in the following order):conda config --env --add channels defaults
conda config --env --add channels bioconda
conda config --env --add channels conda-forge
conda activate paramecium
Install dependencies
We need to install samtools.conda install -c bioconda samtoolsThen install JBrowse2:conda install -c bioconda jbrowse2If you have difficulties installing jbrowse2 from bioconda you can also install npm and then do it manually: https://jbrowse.org/jb2/docs/quickstart_web/
Install GenomeTools so we can add the genome annotation to JBrowse2.conda install -c bioconda genometools-genometools
We also need to download the Paramecium reference genome annotations.curl http://ftp.ensemblgenomes.org/pub/protists/release-55/gff3/paramecium_tetraurelia/Paramecium_tetraurelia.ASM16542v1.55.gff3.gz --output ASM16542v1.55.gff3.gz
Download paired-end fastq data from European Nucleotide Archive, first read 1:curl ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR182/001/ERR1827401/ERR1827401_1.fastq.gz --output read1.fastq.gz
Then read 2:curl ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR182/001/ERR1827401/ERR1827401_2.fastq.gz --output read2.fastq.gz
Perform paired-end alignment
Run alignment with bowtie2:bowtie2 -x paramecium -1 read1.fastq.gz -2 read2.fastq.gz -S alignment.sam
This will align using the paramecium index and perform paired-end alignment with read1 and read2 and then save the output into alignment.sam.
Next, convert the SAM file to a binary BAM file:samtools view -S -b alignment.sam > alignment.bam
Doing anything meaningful such as calling variants or visualizing alignments requires that the BAM is further manipulated. It must be sorted such that the alignments occur in “genome order”. That is, ordered positionally based upon their alignment coordinates on each chromosome.samtools sort alignment.bam -o alignment.sorted.bam
Next, lets index the sorted alignment:samtools index alignment.sorted.bam
Create a JBrowse2 local server
Make a new directory called dev:mkdir dev
cd dev
Then create JBrowse2 inside of this development directory:jbrowse create jbrowse2
cd jbrowse2
Start the server:npx serve .
Open your browser http://localhost:3000/ and you will see something like this:
Sample JBrowse config:
- Volvox sample data
To cancel the server running press CTRL + C while having the terminal window open.
Next lets create an index fasta file (.fai) using samtools. You should have your paramecium fasta (📄ASM16542v1.dna.toplevel.fa) file in the directory structure:
📂paramecium_playground
┣ 📄ASM16542v1.55.gff3.gz
┣ 📄ASM16542v1.dna.toplevel.fa
┣ 📜paramecium.1.bt2
┣ 📜paramecium.2.bt2
┣ 📜paramecium.3.bt2
┣ 📜paramecium.4.bt2
┣ 📜paramecium.rev.1.bt2
┣ 📜paramecium.rev.2.bt2
┗ 📂dev
┗ 📂jbrowse2
Since you are standing in the jbrowse2 directory and the genome (ASM16542v1.dna.toplevel.fa) is two levels up we can call it using ../../ASM16542v1.dna.toplevel.fa :samtools faidx ../../ASM16542v1.dna.toplevel.faWhich will output:
jbrowse add-assembly ../../ASM16542v1.dna.toplevel.fa --load copy --out .Added assembly "ASM16542v1.dna.toplevel" to ./config.json
Next, we will add the BAM file from the alignment:jbrowse add-track ../../alignment.sorted.bam --load copy --out .Which will give the following output:Added track with name "alignment.sorted" and trackId "alignment.sorted" to ./config.json
Add the genome annotation file by doing the following using GenomeTools (gt):gt gff3 -sortlines -tidy -retainids ../../ASM16542v1.55.gff3.gz > ../../ASM16542v1.55.sorted.gff3We should now see the output:
bgzip ../../ASM16542v1.55.sorted.gff3
tabix ../../ASM16542v1.55.sorted.gff3.gz
jbrowse add-track ../../ASM16542v1.55.sorted.gff3.gz --load copyAdded track with name "ASM16542v1.55.sorted.gff3" and trackId "ASM16542v1.55.sorted.gff3" to ./config.jsonThe final step of loading your JBrowse instance may include adding a "search index" so that you can search by genes or other features by their name or ID.jbrowse text-index --out .
Lets now rerun the server now that both alignments and reference genome has been added:npx serve .
Browse the alignments
If you now open your browser http://localhost:3000/ you will see JBrowse2 start screen, click Linear Genome View in the center:

Open the ASM16542v1.dna.toplevel genome.
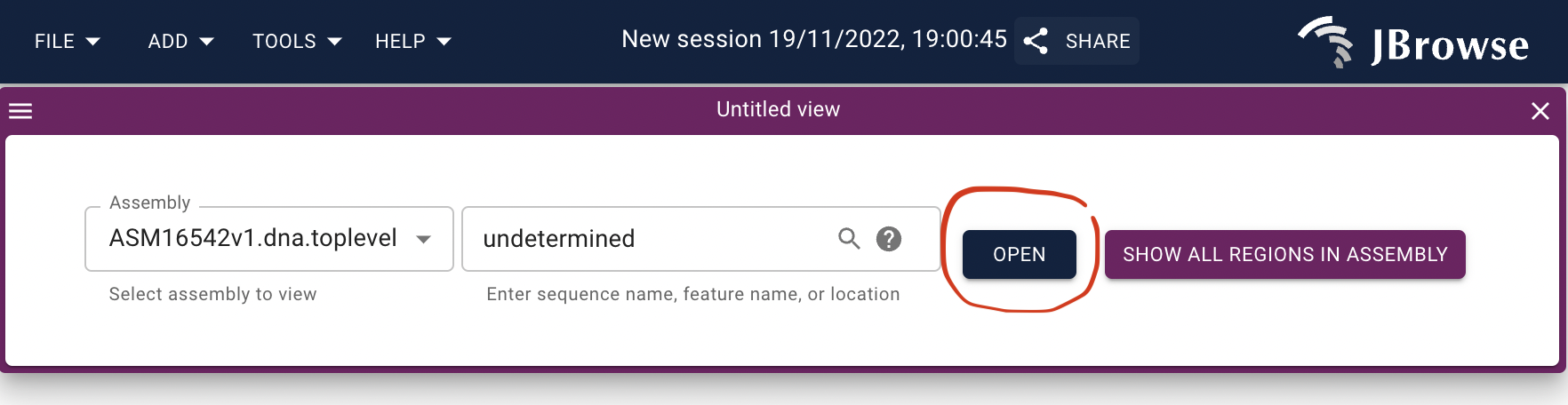
Open track selector.
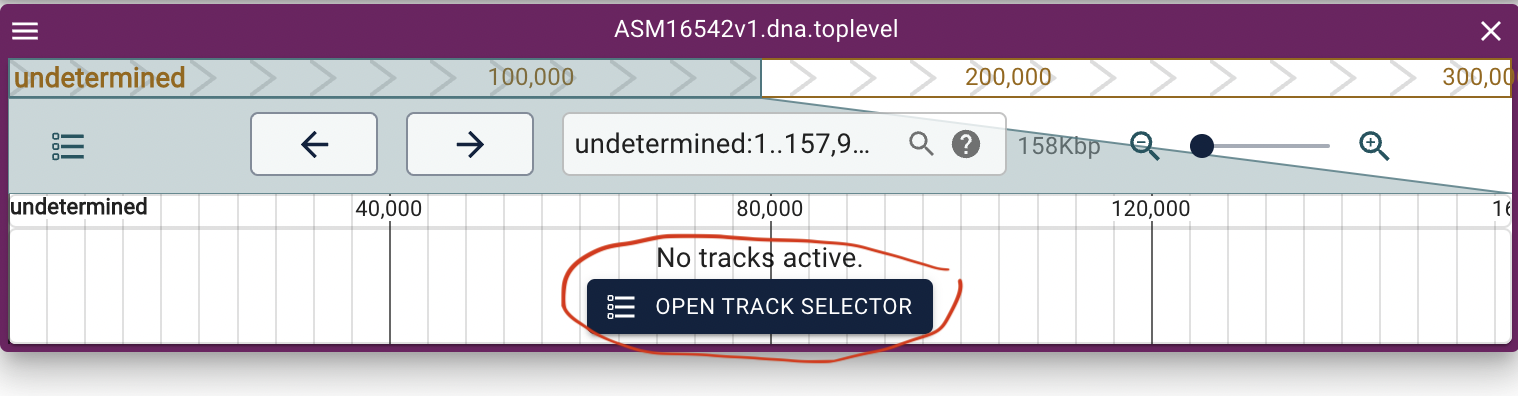
Select to show the annotation track (gff3) and then the alignment track (.bai):

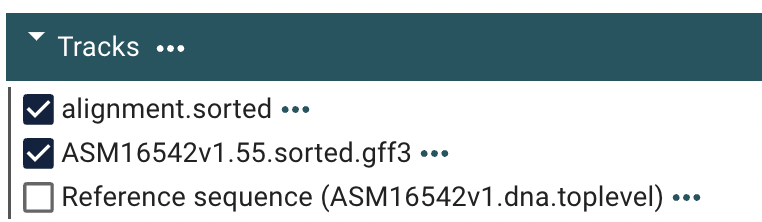
Then lets check a specific gene by searching for it. Search for: GSPATT00027514001.

It will take some seconds for Jbrowse to search. But when it shows up as GSPATT00027514001-1 click on it.
Zoom out a little bit so you can see the entire gene body.
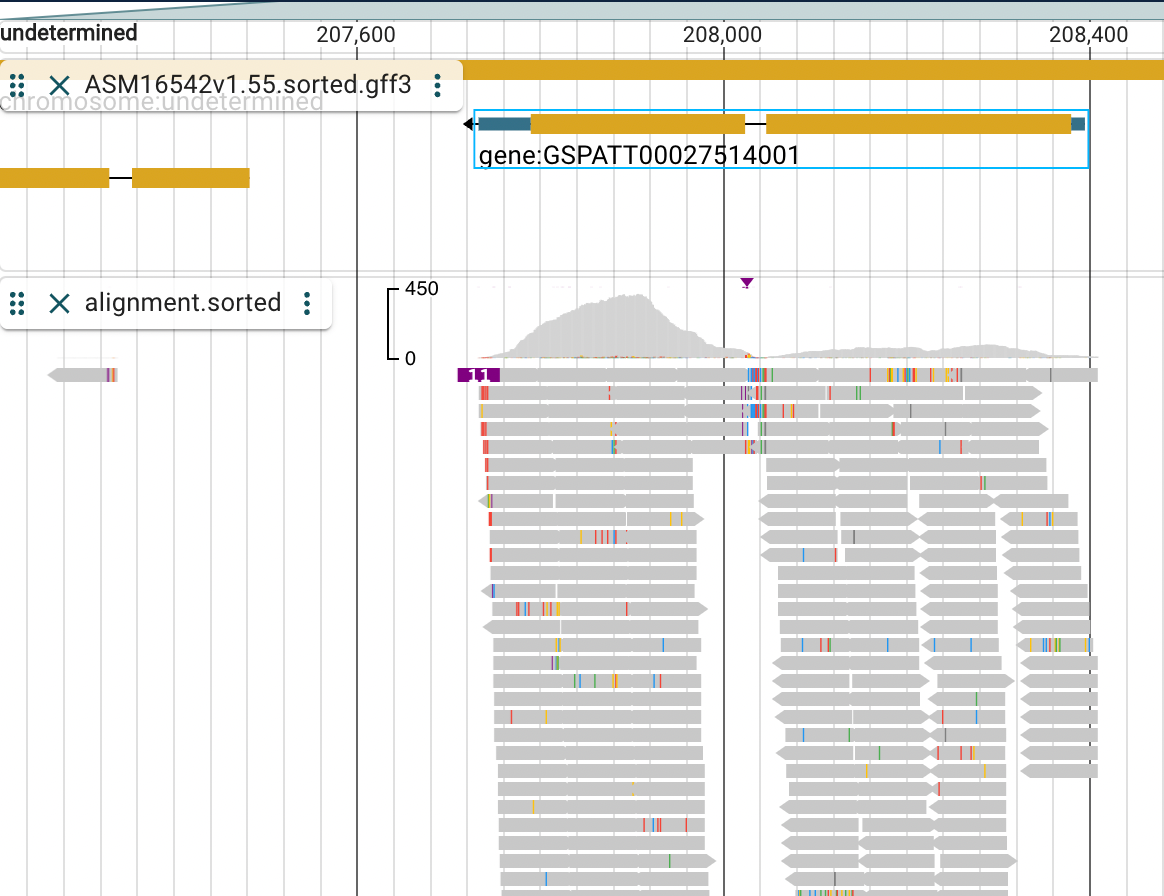
We might ask what gene symbol the gene ID GSPATT00027514001 has? The problem is that the annotation GFF3 file provided by Ensembl does not contain any gene symbols for us to guess its function. So lets use PANTHER, http://www.pantherdb.org/, to search for the gene ID. PANTHER stands for Protein ANalysis THrough Evolutionary Relationships and its purpose is to provide a gene family phylogenetic trees of protein coding genes.

Then search for GSPATT00027514001:

Which will display the following result and we can infact see that the gene ID is equivalent to the translation initiation factor EF1a:
|
|
|
|
|
|||||||||||||||
| 1. | PARTE|EnsemblGenome=GSPATT00027514001|UniProtKB=A0EIP4 | S1-like domain-containing protein GSPATT00027514001 PTN002729134 orthologs |
EUKARYOTIC TRANSLATION INITIATION FACTOR 4C (PTHR21668:SF0) | translation initiation factor | Paramecium tetraurelia | ||||||||||||||
We can also find the gene in the Uniprot database where it has the gene name A0EIP4.
With the advent of Google DeepBrain's AlphaFold we can even see the predicted 3D protein structure:
